|
One of the exciting observations from examining hundreds of bacterial genomes is the enormous amount of diversity, even if the genomes come from the same bacterial species. This diversity is generated by a variety of mechanisms and is responsible for different traits e.g., bacterial pathogenicity and antibiotic resistance.
The huge amount of data available for bacterial genomes provides the opportunity to study exciting questions in the bacterial world using data mining and comparative genomics techniques. In our research group, we find these questions and develop algorithms and pipelines, to address them. |
|
|
Bacterial genome-wide association studies
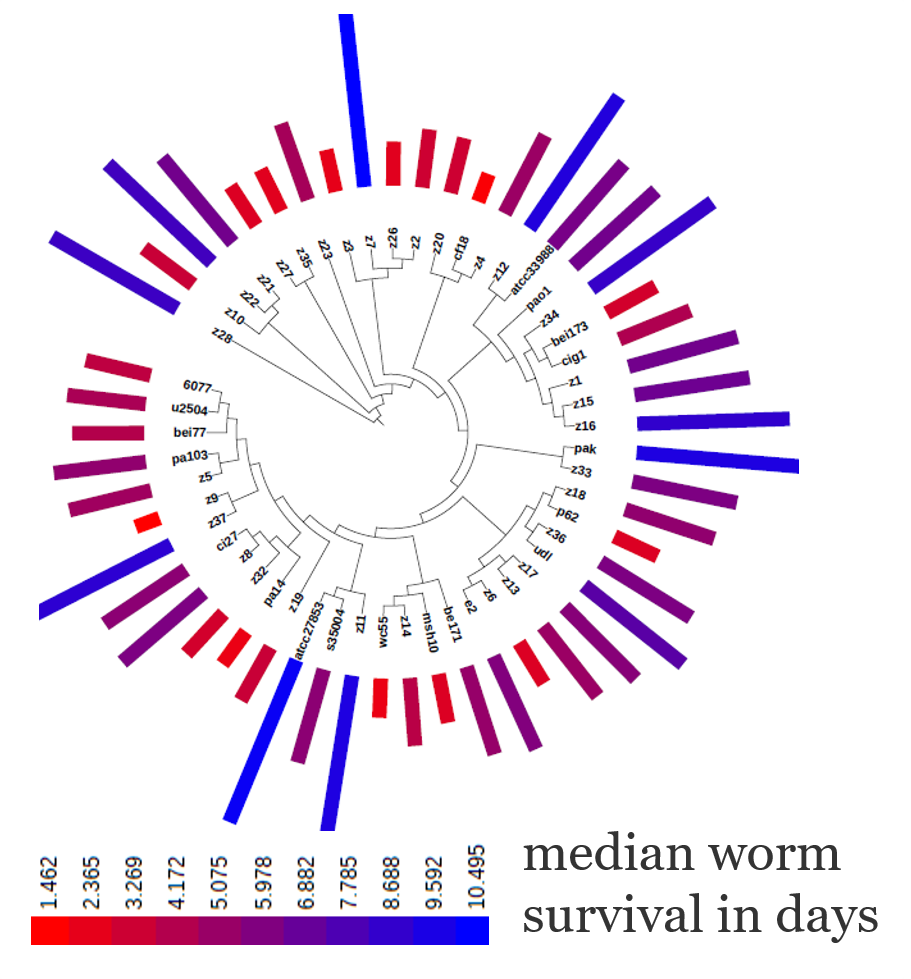
We use comparative genomic approaches to identify correlations between bacterial phenotypes (e.g., virulence towards the host) and the presence/absence of specific genomic elements. In a recent study, we examined 52 P. aeruginosa strains that display a broad range of virulence towards the worm C. elegans, and identified accessory genome elements that correlate with virulence, including both known and novel virulence determinants (Genome Biology, 2019). |
|
Optimization of Bacterial Strain Typing
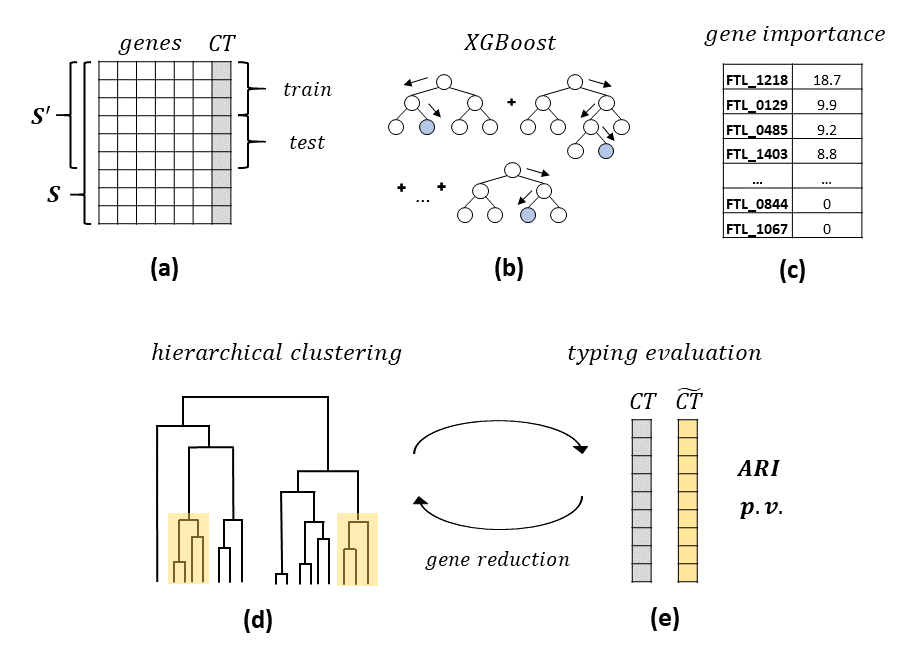
High-resolution microbial strain typing is essential for various clinical purposes. The most widely used gene-by-gene approach is based on multilocus sequence typing (MLST), comprised of typing schemes that utilize a different number of genes, each is suitable for addressing different levels of isolate discrimination. Schemes based on the bacterial core genome (cgMLST) provide a high level of typeability and maximal discriminatory power. We developed a new machine-learning-based methodology, minMLST, for minimizing the number of genes in cgMLST schemes by identifying subsets of informative genes and analyzing the trade-off between gene reduction and typing performance. The optimized MLST schemes are expected to improve inter-laboratory communication (Bioinformatics, 2020). |
|
Gene Block Discovery
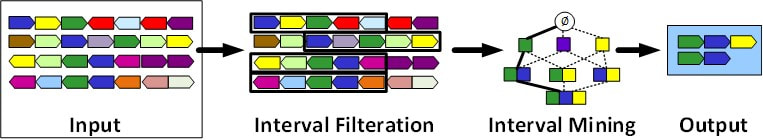
Recent advances in Next Generation Sequencing techniques, combined with global efforts to study infectious diseases, yield huge and rapidly-growing databases of microbial genomes. These big new data statistically empower genomic-context based approaches to functional analysis: the idea is that groups of genes that are clustered locally together across many genomes usually express protein products that interact in the same biological pathway and can be considered “gene blocks”. We addressed a new gene block discovery problem variant: find conserved gene blocks abiding by a user specification of biological functional constraints. We took advantage of the biological constraints to efficiently prune the search space (Journal of Computational Biology, 2019). |
